本文根據理化學研究所成果發布資料編譯而成

日本理化學研究所(以下簡稱「理研」)生命機能科學研究中心計算分子設計研究團隊的小松輝久研究員、衝本憲明高級研究員及泰地真弘人組長等人組成的研究團隊,對新冠病毒感染症(COVID-19)的致病病毒「SARS-CoV-2」的主蛋白酶(Mpro)蛋白與7種人類免疫不全症病毒(HIV)蛋白酶抑制劑結合的過程進行了分子動力學模擬(影片)。
病毒通過使感染的細胞產生病毒蛋白來增殖。SARS-CoV-2的Mpro主要發揮「剪刀」(蛋白酶)的作用,負責在適當的位置剪斷細胞產生的蛋白質並使其變得完整。Mpro與HIV的蛋白酶類似,因此有望利用現有的HIV蛋白酶抑制劑治療SARS-CoV-2。
研究團隊對7種HIV蛋白酶抑制劑分別與Mpro表表面接觸的過程進行了分子動力學模擬,調查了容易結合的位點分類,以及與蛋白酶活性位結合的難易度。另外,通過解析位點的結構變化,發現這種結構的變化程度非常大,即使是在與抑制劑結合的狀態下,也能呈現多種形狀和排列。此次的研究成果驗證了抑制劑與靶蛋白的動態結合過程,為發現候選藥物分子提供了新的可能性。
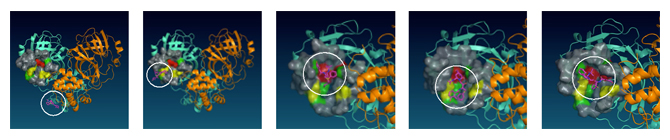
HIV抑制劑(奈非那韋=白色圓圈中的分子)與Mpro結合的過程
研究方法與成果
為推進抑制新冠病毒主蛋白酶(Mpro)的藥物分子開發,研究團隊通過分子動力學計算,模擬了已作為HIV抑制劑使用的7種藥物分子與Mpro結合的過程。分子動力學計算利用了理研資訊基礎中心的HOKUSAI系統和專用計算機MDGRAPE-4A執行。
SARS-CoV-2 Mpro的空間結構根據Liu等人的報告注1)設定,作為溶劑的水分子數量接近3萬個,系統的總原子數量接近10萬個。將7種藥物分子分別添加到含Mpro的水溶液中,並逐一對添加了不同藥物分子的系統實施0.2微秒(1微秒為100萬分之1秒)×28次的模擬,觀察藥物分子在Mpro表面的附著情況。然後,根據通過全部試驗(7×28次)數據獲得的藥物分子與Mpro的接觸數據,對Mpro表面藥物容易粘附的位置(結合位置)進行了分類,並確定其中一個位置位於與Mpro的剪刀功能直接相關的位點(蛋白酶活性位,圖1)。另外,通過調查0.2微秒的結合過程,還確認了藥物分子與活性位實際結合的情形(圖2)。
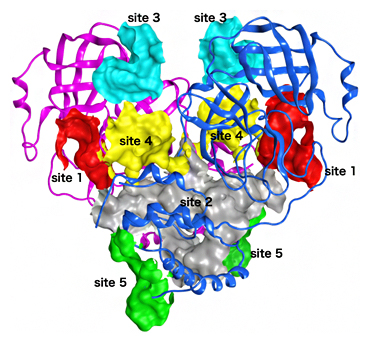
圖1:SARS-CoV-2主蛋白酶(Mpro)的結合位置
Mpro作為由兩個蛋白質次單元組成的同型二聚體發揮作用。根據7種藥物分子的結合數據,對Mpro表面的結合位置進行了分類,顯示了5個主要位點(site1-site5)。用紅色顯示的site1與蛋白酶活性位重合。
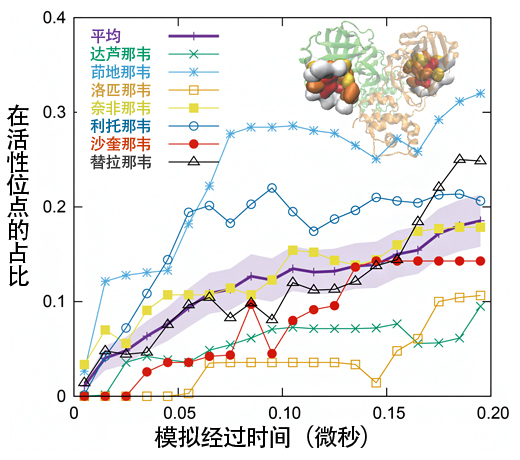
圖2:藥物分子與活性位結合的過程
本圖顯示了在利用7種藥物分子的MD模擬中,各藥物分子在蛋白酶活性位的佔比隨時間發生的變化。可以看出,在0.2微秒的時間裏,與活性位的結合逐漸增加。受樣品數量限制,很難準確預測每種藥物分子的優劣,但茚地那韋(淡藍色)的親和性明顯高於達蘆那韋(綠色)。
為捕捉與藥物分子結合的Mpro的結構變化,針對在上述模擬中各藥物分子最終與活性位結合的23次試驗延長了分子動力學計算,觀察了1微秒内的動態。由此發現,結合位置的形狀會大幅變化。另外,將3次試驗的計算時間延長至6微秒後,偶爾會觀察到藥物分子在結合位置中改變方向(圖3)。這表明,藥物分子與靶蛋白的結合比以前認為的要靈活得多。
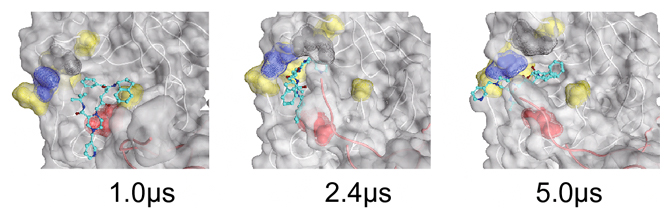
圖3:在結合位置中改變方向的藥物分子
對茚地那韋(用淡藍色主鏈顯示的棍狀模型)與結合位置site-1(活性位)的結合進行6微秒MD模擬的結果。茚地那韋的方向在1.0微秒(μs)、2.4微秒和5.0微秒時大幅改變。Mpro用空間填充模型表示,與跟茚地那韋結合有關的胺基酸殘基分別用紅色(第166位麩胺酸)、藍色(第189位麩醯胺)、黃色(第44位半胱胺酸、第143位甘胺酸、第187位天冬氨酸、第188位精胺酸、第190位蘇胺酸)和灰色(第49位甲硫胺酸)顯示。可以看出,這些胺基酸殘基的位置關係也在發生變化。
未來展望
今後,為獲得用於探索候選藥物分子和開發新藥物分子的數據,研究團隊預定利用更廣泛的藥物分子,對在SARS-CoV-2 Mpro和SARS-CoV-2中編碼的其他潛在新藥靶蛋白進行模擬。
注1)Title: The crystal structure of COVID-19 main protease in complex with an inhibitor N3, Entry authors: Liu, X., Zhang, B., Jin, Z., Yang, H., Rao, Z., Initial deposition on: 26 January 2020
論文資訊
題目:Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation
期刊:Scientific reports
DOI:10.1038/s41598-020-74099-5
成果發布資料
編譯:JST客觀日本編輯部