本文根據日本東北大學成果發布資料編譯整理而成

日本東北大學加齡醫學研究所基因表現控制領域的岡崎慶鬥助教、關根弘樹講師和本橋Hozumi教授組成的研究團隊,與該研究所呼吸外科學領域的岡田克典教授、東北大學醫學系研究科的鈴木貴教授,以及該校資訊科學研究科和Medical Megabank Organization的木下賢吾教授等人合作,共同在NRF2被激活的癌症(以下稱「NRF2激活癌」)中發現了維持癌症幹細胞乾性所需的基因體區域。研究發現,該基因體區域通過在NRF2激活癌中發揮獨特性功能並增加NOTCH3蛋白來維持癌症幹細胞乾性。這項研究成果有望用來有效治療對抗癌藥產生抗藥性的NRF2激活癌。
背景
轉錄因子NRF2是通過與名為抗氧化響應序列的鹼基序列結合來激活轉錄的蛋白質。在常態下,NRF2通過激活參與生物防禦的各種基因來進行解毒代謝和響應氧化壓力,對維持我們的健康起著重要作用(圖1左)。另一方面,已知在肺癌和頭頸癌中,NRF2會被特定的基因突變異常激活,導致病情惡化(圖1右)。
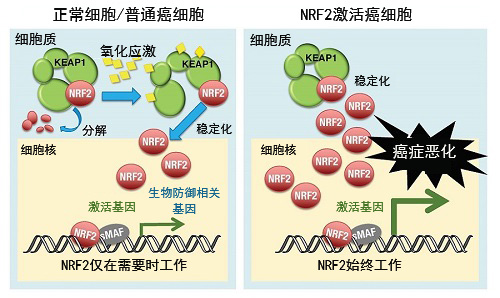
圖1:在NRF2激活癌中,NRF2始終發揮作用,導致癌細胞惡化
在正常細胞和普通癌細胞中,轉錄因子NRF2的功能受細胞質蛋白KEAP1調控(左圖)。平時KEAP1會誘導NRF2分解,抑制NRF2的功能。細胞受到氧化壓力後,KEAP1便不再發揮作用,NRF2變得穩定,並發揮轉錄因子的功能,一舉激活參與生物防禦的基因簇。而在部分癌細胞中,KEAP1對NRF2的分解出現問題,NRF2始終保持穩定。這種NRF2激活癌細胞的致瘤性很強,對抗癌治療的耐性也比較強,屬於難治性癌症。
此前的研究表明,抑制被異常激活的NRF2的功能可以抑制這類癌細胞增殖,改善抗癌藥的效果。不過,服用NRF2抑制劑的話,全身的正常細胞的NRF2也會被抑制,考慮到生物防禦因子NRF2的重要性,這樣可能會發生各種副作用。因此,需要開發能把對正常細胞的影響降到最低,同時可以有效滅絕NRF2激活癌的治療靶標。
此次的研究
研究團隊在NRF2被特定基因突變異常激活的NRF2激活肺癌細胞和普通肺癌細胞中比較了NRF2的工作方式。結合名為RNA序列和ChIP序列的綜合分析方法比較NRF2控制的基因表現和增味劑的形成發現,NRF2的工作方式存在差異。也就是說,此次發現,在NRF2激活肺癌細胞中,NRF2通過與另一個轉錄因子CEBPB統合工作,來與異常的基因座結合形成增味劑,並激活該基因的轉錄(圖2)。尤其是發現,NRF2與NOTCH3基因座結合的基因體區域會作為增味劑增加NOTCH3蛋白,從而增強癌症幹細胞的乾性。這意味著,抑制NOTCH3的話,可以抑制惡性程度較高的NRF2激活肺癌的癌症幹細胞乾性。
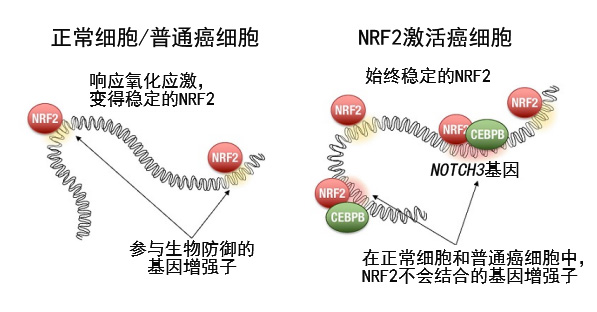
圖2:在NRF2激活癌中,NRF2通過與平時不會結合的位點結合來形成增味劑
調查基因體上的NRF2結合位置發現,NRF2在NRF2激活癌細胞中的結合位置與正常細胞和普通癌細胞不同。NRF2通過與另一個轉錄因子CEBPB統合工作,可以在NRF2激活癌中作用於特徵性基因座。其中之一是NOTCH3基因座,會隨著與NRF2結合形成增味劑,蠻力激活NOTCH3的基因表現。NOTCH3能增強NRF2激活癌細胞的幹細胞乾性,加劇癌症惡化。
研究的意義
在NRF2激活癌中,代謝細胞膜上的藥物的轉運蛋白表達增強,抗癌藥會快速代謝掉,這被認為是此類癌症難以治療的原因之一。NOTCH3為膜蛋白,可以從細胞外抑制其功能,因此靶向NOTCH3的治療戰略被認為可以避免NRF2激活肺癌中增強的藥物代謝轉運蛋白的影響(圖3)。研究團隊已經通過動物實驗發現,與NOTCH3胞外域發生反應的抗體對NRF2激活癌有抗腫瘤效果、在通過抑制NOTCH3來抑制癌症幹細胞乾性的同時使用細胞毒性抗癌藥可以發揮互利共生。
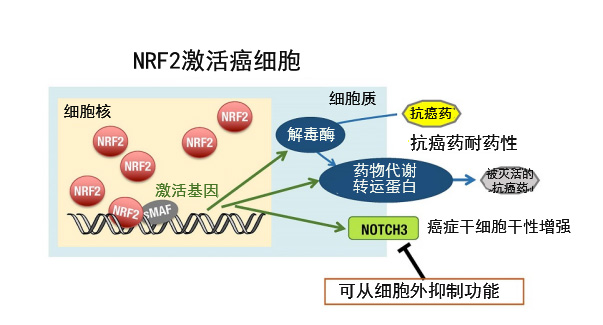
圖3:對於能蠻力解毒代謝抗癌藥物的NRF2激活癌,可以從細胞外抑制功能的NOTCH3是有效的治療靶點
NRF2激活癌細胞中高度表達藥物的解毒酶和向細胞外代謝藥物的轉運蛋白,對抗癌藥的抗藥性增強。NOTCH3為膜蛋白,可以從細胞外抑制其功能。因此,可以說NOTCH3是治療NRF2激活癌的有效靶點。
此次研究通過明確NRF2激活癌中的特徵性NRF2的功能,確定了維持癌症幹細胞乾性所需的基因體區域。另外還發現,在該區域的作用下產生的NOTCH3蛋白可作為NRF2激活癌這種難治性癌症的新的有效治療靶點。NRF2激活癌可通過分析癌症的基因突變診斷,因此,此次的研究成果有望成為根據癌症的基因診斷為患者量身定製治療方案的先驅研究成果。
論文資訊
題目:Enhancer Remodeling Promotes Tumor-Initiating Activity in NRF2-Activated Nonsmall Cell Lung Cancers
期刊:Nature Communications
DOI:10.1038/s41467-020-19593-0
日語發布
編譯:JST客觀日本編輯部