日本東北大學研究生院藥學研究科的中林孝和教授、田原進也助教及研究生山崎公介等人組成的研究團隊,明確了與肌萎縮性側索硬化症(ALS)發病有關的SOD1毒性是如何在分子層面獲得的新分子機制。
ALS是一種進行性神經退行性疾病,發病原因不明,所以治療方法始終沒有進展。雖然研究人員在ALS的病灶部位發現了SOD1(銅離子與鋅離子相結合的金屬蛋白)聚集物,但SOD1毒性與ALS發病之間的關係,此前一直不清楚其詳細機制。
此前研究團隊證明,當SOD1的結構發生變化時,不但沒有抗氧化作用,反而是具有氧化周圍化合物的高毒性氧化作用,對此現象,研究人員研究了其氧化機制。此次首次成功證明,SOD1的抗氧化作用和氧化作用這兩種完全相反的功能是通過SOD1内的單個硫-硫鍵(雙硫键)的生成和斷裂來轉換抗氧化和氧化作用的。
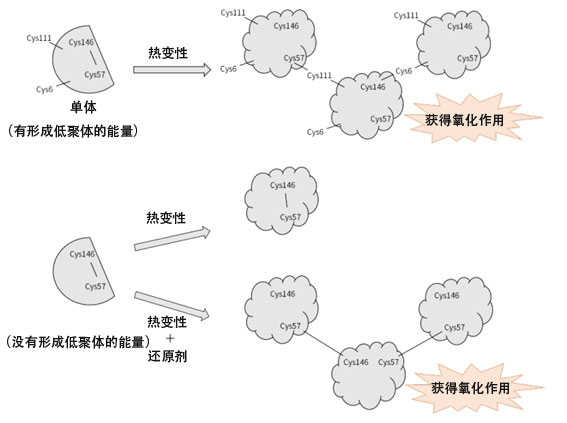
圖1:隨著SOD1低聚體化發生的分子内雙硫键的斷裂及獲得氧化作用的模式圖。隨著形成低聚體,分子内雙硫键斷裂,獲得氧化作用(供圖:東北大學研究生院藥學研究科中林孝和教授)
SOD1表達細胞毒性與SOD1聚集體無關,而是與數個SOD1分子聚集形成的低聚體有關。研究團隊發現,硫-硫鍵斷裂會形成低聚體,這種低聚體具有強氧化作用。由此提出了SOD1低聚體的毒性源自氧化作用的觀點。
為驗證這一點,研究團隊準備了具有低聚體形成能力的突變體和不具備這種能力的突變體。SOD1通過半胱胺酸殘基(Cys)之間的分子間雙硫键形成低聚體。SOD1有4個Cys,Cys6和Cys111作為還原型遊離硫醇基(-SH基)存在,Cys57和Cys146在分子内形成雙硫键。
研究團隊針對Cys6和Cys111置換了遊離Cys,消除了分子間的雙硫键形成能力,製備了不會形成低聚體的突變體。利用形成低聚體的普通SOD1和不會形成低聚體的突變體,研究了熱變性引起的低聚體化與氧化作用的關係。利用DCF螢光法對熱變性之前和之後的氧化作用進行量化發現,具有低聚體形成能力的普通SOD1發生熱變性後,觀測到了低聚體的形成和氧化作用的增大。而置換了遊離Cys的突變體不僅沒有觀測到低聚體的形成,也沒有發生氧化作用。
這些結果表明,低聚體的毒性是形成低聚體時分子内雙硫键斷裂引起的氧化作用所致。獲得氧化作用不需要熱變性和形成低聚體,僅通過切斷分子内的雙硫键即可獲得。此次研究發現,僅僅是分子内的雙硫键斷裂,SOD1的酶活性就會從保護身體的抗氧化作用轉變成有毒的氧化作用。
中林教授表示:「此次的研究表明,SOD1的分子内雙硫键斷裂會將酶活性由抗氧化作用轉變為氧化作用。通過靶向分子内雙硫键,有望從基礎科學方面開發應用於治療和預防的藥物。」
【詞注】
■DCF螢光法:利用二氯螢光素(DCF)分子的螢光強度評估活性氧生成量的方法。
原文:《科學新聞》
翻譯編輯:JST客觀日本編輯部
【論文資訊】
期刊:Scientific Reports
論文:SOD1 gains pro-oxidant activity upon aberrant oligomerization: change in enzymatic activity by intramolecular disulfide bond cleavage
DOI:10.1038/s41598-022-15701-w