日本理化學研究所生命醫科學研究中心基因體解析應用研究團隊的小杉俊一研究員(現靜岡縣立綜合醫院研究支持中心遺傳研究部研究員)、寺尾知可史組長(靜岡縣立綜合醫院臨床研究部免疫研究部長)等人組成的共通研究團隊發表研究成果稱,開發出了一種在全基因體測序數據中高精度檢測結構變異 (Structure Variantions,簡稱SV) 的新方法。相關成果已刊登在《Cell Genomics》上。
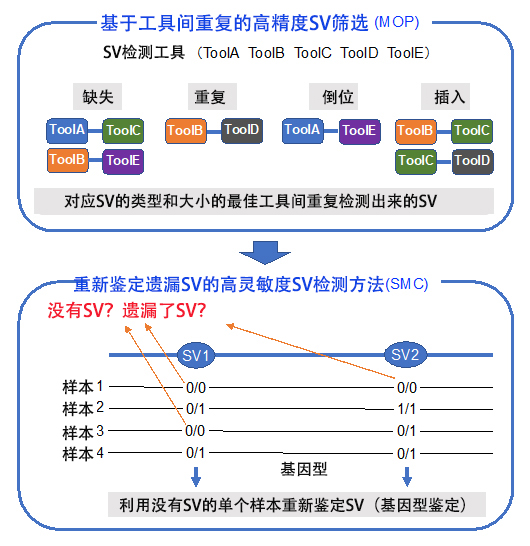
圖1 本次開發的「MOPline」演算法(供圖:理化學研究所)
基因體的SV是超過50個鹼基對(bp)的缺失、插入、重複、倒位變異的統稱,有別於小於50bp的缺失、插入對應的插入缺失(indel),以及單個鹼基置換的單鹼基變異(SNV)。大量研究表明,導致個體基因體之間存在巨大差異的SV是包括發育障礙和智力障礙在内的各種人類疾病和表徵的遺傳因素。
另一方面,由於SV結構複雜和尺寸較大,其檢測比SNV更加困難。基因體多態性通常通過將100~150bp的短序列(擷取)數據與標準人類基因體序列(參考序列)進行對齊來檢測。雖然SNV和插入缺失可納入擷取長度内,但尺寸較大的SV無法納入擷取盒内,所以必須使用跨SV對齊擷取的間接證據檢測,從而導致檢測精度和靈敏度降低。
儘管目前已經開發出多種SV檢測工具,但一直存在檢測結果通用性較低的問題,還沒有能夠通過單一工具實現高精度和高靈敏度SV檢測的工具。
研究團隊發現,能夠進行高精度檢測的重疊SV(現有SV檢測工具間共同檢測出的SV)並沒有表現出高精度,所以研究團隊調查了現有工具的最佳組合。然後,開發了使用現有的4~9個工具按SV類型和大小決定最佳工具組合的演算法,並將其命名為MOP(Merging Overlap calls from selected Pairs of algorithms)。
若使用MOP,雖然可以鑑定具有高精度的SV,但會漏掉一部分SV。因此,通過掃描用MOP未能檢測出SV的基因體區域,確認了SV的存在。在這一存在確認中,使用了一種利用擷取對齊資訊的獨特基因型鑑定方法,這種重新鑑定SV的方法被命名為SMC(Supplementing Missing Calls)。
最終,研究團隊成功開發出結合MOP、SMC、過濾和標註功能的SV檢測方法——MOPline。
使用全基因體測序數據驗證MOPline的SV檢測精度和靈敏度時,顯示其結果超過了現有工具的精度和靈敏度。此外,使用從公共數據庫(千人基因體計劃)獲得的100個全基因體測序數據,將多個工具組合與檢測SV的現有管道進行了比較。結果顯示,MOPline的SV檢測精度與現有工具相當,但檢測到的真陽性SV(尤其是插入)的數量(檢測靈敏度)超過了現有工具。
其次,使用MOPline從3258人的BBJ全基因體測序數據中檢測SV後發現,檢測到出大約13萬4000(個人平均約1萬6000)的SV,這一數字比迄今為止的大規模SV研究項目中檢測到的個人平均SV數量高出1.7~3.3倍。
該BBJ全基因體測序數據來自至少患有癌症和癡呆症等中的一種疾病的患者。因此,對與疾病相關的已知基因的蛋白質編碼區域重疊的SV進行調查後發現,幾種罕見的SV與疾病樣本中特異的已知疾病風險基因(大腸癌、乳房癌等)的蛋白質編碼區域重疊。
以MOPline檢測到的BBJ-SV為參照面板,對18萬人的SNP陣列數據(SNP基因型數據)進行填補,推斷出約18萬人的SV。使用推斷的SV和約18萬人的醫療資訊,對42種疾病和60種定量性狀進行了全基因體關聯分析(GWAS)後發現,針對包括癌症等疾病在内的32種表徵,發現了41個與SNP具有同等或更強相關的SV。在相關的SV中,8個SV與相關基因的編碼區重疊,其中5個是之前未報導與該表徵相關的新鑑定的SV。
MOPline是一種展示了前所未有的SV檢測精度和靈敏度的工具,不僅可以鑑定導致單基因疾病的罕見SV,還可以通過對SV進行填補,鑑定與數量表徵相關的SV。
原文:《科學新聞》
翻譯:JST客觀日本編輯部
【論文資訊】
雜誌:Cell Genomics
論文:Detection of trait-associated structural variations using short read sequencing
DOI:10.1016/j.xgen.2023.100328