以日本近畿大學醫學部内科學教室的渡邊智裕特命教授和研究生院醫學研究科的博士生大塚康生為核心的研究團隊發布研究報告稱,查明瞭因真菌感染引發免疫反應,從而激活細胞内分子的富白胺酸重複激酶(LRRK2),導致急性胰腺炎重症化的機制。相關成果刊登於《Clinical and Experimental Immunology》上。
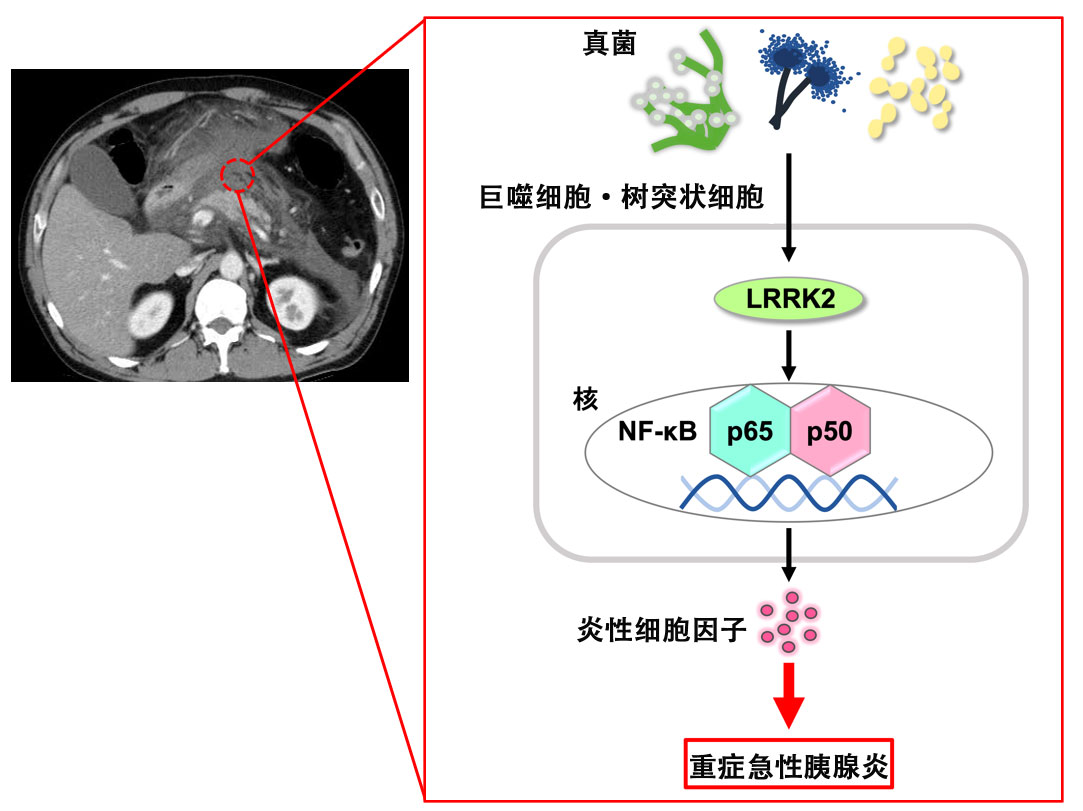
圖1 重症急性胰腺炎的發病機製(供圖:近畿大學)
急性胰腺炎患者數量在不斷增加(2016年為7.8萬人),其中19.7%發展為重症,重症死亡率約為20%。據分析,酒精或高脂肪飲食會激活胰腺腺泡細胞中的胰蛋白酶原,導致消化酶胰蛋白酶過量產生,促進自溶,從而導致急性胰腺炎惡化,但詳細機制尚未明確,也沒有確立根治療法。此外,由於缺乏胰酶的小鼠也會發生胰腺炎,因此研究人員認為該疾病的發生可能還涉及其他機制。該研究團隊先前已經證明,NOD1介導的對腸道細菌的免疫反應會加重胰腺炎。
博士生大塚表示:「重症急性胰腺炎的3~4成會並發真菌感染,有研究統計這類病例的死亡率高達40%。此外,LRRK2雖是巴金森氏症易感蛋白之一,但也被認為參與了對真菌的免疫反應,因此我們重點研究了LRRK2。」
研究團隊首先給體內給與誘發急性胰腺炎藥物的小鼠投用了LRRK2抑制劑。結果顯示,抑制劑阻止了免疫細胞向胰腺的浸潤,並減少了胰腺中炎性細胞介素的產生,從而抑制了胰腺炎。
此外,當對過度表達LRRK2的小鼠投用藥物誘發急性胰腺炎後,發現胰腺炎惡化。過度表達小鼠中浸潤胰腺的巨噬細胞增加,並且發炎細胞介素的產生也有所增加。此時,LRRK2在與胰腺炎相關的胰腺腺泡細胞和免疫細胞中都有表達。
於是,研究人員建立了僅在免疫細胞中過度表達LRRK2的小鼠並誘發了急性胰腺炎,結果發現急性胰腺炎惡化,並且免疫細胞也參與了惡化。LRRK2通過激活轉錄因子NF-κB,增加了炎性細胞介素IL-6、TNF-α和IL-12/23p40的產生,導致胰腺炎加重。
那麼究竟是什麼激活了LRRK2呢?研究人員嘗試給過度表達LRRK2的小鼠投餵含有抗細菌藥的水以清除腸道細菌,發現胰腺炎並未受到抑制。另一方面,當投餵含有抗真菌藥的水清除腸道真菌時,發現胰腺炎受到抑制,炎性細胞介素也顯著減少。
正在推進巴金森氏症臨床試驗
渡邊特命教授表示:「首先,急性胰腺炎會破壞胰腺屏障,使腸道真菌侵入胰腺,當免疫細胞識別到這一點時,LRRK2就會被激活。我們認為被激活的LRRK2會通過產生炎性細胞介素使胰腺炎加重。」
本次研究揭示了通過LRRK2的功能調節和消除腸道真菌,創造新的急性胰腺炎治療方法的可能性。此外,LRRK2抑制劑在巴金森氏症等中的臨床試驗正在推進中。
渡邊特命教授表示:「雖然很難量化LRRK2,但目前已知基因突變與巴金森氏症、克羅恩病、痲瘋病等有關。通過檢查血細胞的基因序列,或許可以預測疾病的重症化。在此基礎上,可以考慮使用抑制劑等來防止重症化。」
原文:《科學新聞》
翻譯:JST客觀日本編輯部
【論文資訊】
雜誌:Clinical and Experimental Immunology
論文:Leucine-rich repeat kinase 2 promotes the development of severe experimental acute pancreatitis
DOI:10.1093/cei/uxad106