由名古屋市立大學研究生院醫學研究科的研究員中村勇治、講師嶋田逸誠、教授加藤洋一、齋藤伸治教授等組成的研究團隊發表研究成果稱,在與名古屋大學研究生院醫學研究科特任教授尾崎紀夫、特任講師有岡祐子、以及東京大學、京都大學、熊本大學等共同開展的12國國際合作研究中,成功利用類腦器官明確了小兒神經疑難病的病理機制,發現了導致人類神經幹細胞減少的責任基因。這項研究是通過結合基因分析和三維培養模型實現的,將有助於開發新的治療方法。相關研究成果已於7月31日刊登在國際學術期刊《Brain》上。
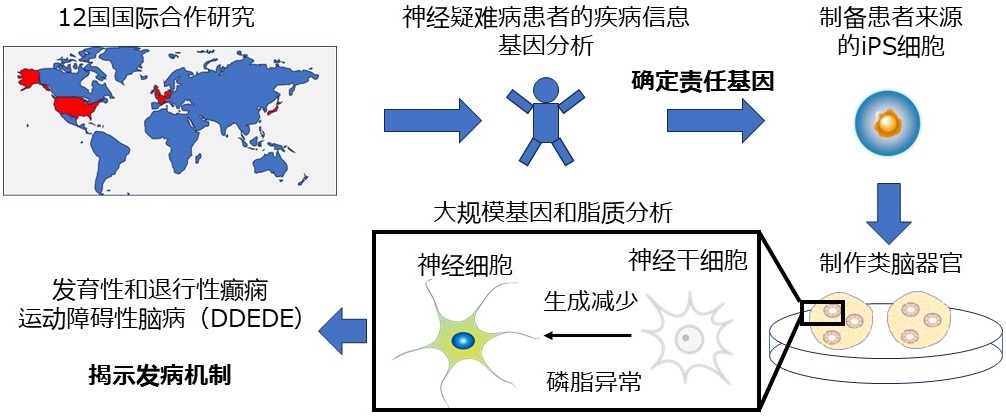
研究概要圖(供圖:名古屋市立大學)
小兒罕見病中存在大量難以診斷的疾病,即使進行全基因體分析也很難發現致病基因並明確其影響。
此次,研究團隊分析了伴有發育性和退行性癲癇運動障礙的神經疑難病患者的基因,在磷脂分解酶「磷脂酶A2」成員之一的PNPLA8基因上發現了突變。
隨後,研究團隊利用iPS細胞製作了三維腦培養模型(類腦類器官模型),研究了PNPLA8基因功能失落所造成的影響。
結果發現,PNPLA8基因的功能失落會導致人類獨特性神經幹細胞變少,大腦尺寸變小。研究表明,根據PNPLA8基因功能失落的程度,該病會由輕症型向重症型轉變,重症型大腦將無法形成褶皺導致小頭症,輕症型會出現行走困難等症狀。
研究揭示了PNPLA8基因是導致發育性和退行性癲癇運動障礙性腦病(命名為DDEDE)的責任基因。
通過使用PNPLA8基因缺失的iPS細胞和患者來源的iPS細胞製造腦類器官,研究人員還發現,被稱為「外側放射狀膠質細胞」的人類獨特性神經細胞數量出現減少,進而導致神經生成減少。
此外,研究團隊通過大規模基因和脂質分析發現,磷脂代謝的異常可能導致神經幹細胞無法正常分化,從而對神經生成產生影響。研究還證實,當添加了缺乏的磷脂之後,人類獨特性神經幹細胞的數量有所恢復。
據悉,研究團隊今後將結合各種動物模型,進一步闡明兵力機制並開發治療方法。
齋藤教授表示:「由於本次研究的對象是一類極其罕見的疾病,為了集中患者資源,我們通過一項由12個國家的63名研究人員參與的國際合作研究實施了這項工作。此外,在日本進行的功能解析是與日本國内的8所大學合作開展的。我們切身感受到,國際合作研究對於研究罕見遺傳疾病是必不可少的,同時我強烈感受到,高水平的研究需要與擁有高超技術的研究人員的共同合作。」
原文:《科學新聞》
翻譯:JST客觀日本編輯部
【論文資訊】
期刊:Brain
論文:Biallelic null variants in PNPLA8 cause microcephaly by reducing the number of basal radial glia
DOI:doi.org/10.1093/brain/awae185