筑波大學人類綜合科學研究科生命系統醫學專業的坂本和雄(現為神戶大學特定助教)與川西邦夫助教(現為昭和醫科大學教授)、筑波大學生存動力學研究中心的金俊達助教(現為富山大學副教授)、東海大學醫學部的松阪泰二教授等人的研究團隊宣佈,已解明左右腎臟試圖維持功能性與結構性平衡現象(腎臟相互平衡)的分子機制。該發現是通過開發並分析能在左右腎臟中僅單側誘導損傷的小鼠模型而得出的結論。研究表明,血管收縮素Ⅱ(AngII)的非對稱局部激活可能是腎臟相互平衡的成因之一。該成果有望推動腎臟疾病治療方法的開發。相關研究成果已發表在期刊《日本學士院紀要B》6月20日刊上。
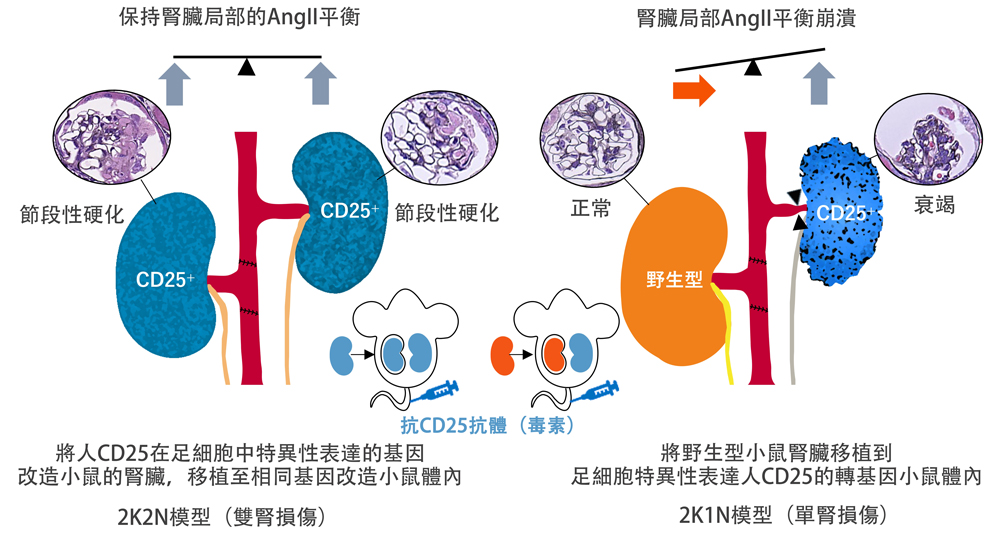
圖1 研究成果的概要(提供:神戶大學)
腎臟是一對左右成對的器官,當一側腎臟受損時,受損腎臟會逐漸出現血流下降和結構性萎縮,而對側腎臟會表現出代償性的功能亢進和肥大。這種左右腎臟之間的動態平衡調節被稱為腎臟相互平衡。但其機制長期以來未被闡明。
慢性腎臟病由老化、高血壓、糖尿病等多種主要因素引發,病情的進展會導致腎功能不可逆地下降,進而需要透析或腎移植。全球的慢性腎病患者呈增加趨勢,近年來,早期診斷和干預的重要性逐漸得到重視。
在腎臟疾病的研究中,以往一直使用對單側腎小管間質施加損傷以再現疾病的動物模型。但這種模型在研究由腎小球引起的病灶,以及將左右腎臟視為可能相互影響的系統的腎臟疾病方面存在侷限。
為此,本次研究團隊製作了向小鼠腎小球細胞施加選擇性損傷、且僅在單側腎臟誘導足細胞(腎臟腎小球表面的上皮細胞)損傷的2K1N模型。
具體而言,研究人員使用腎小球足細胞特異性地表達人CD25的NEP25小鼠(腎病症候群模型),通過施用針對CD25的免疫毒素LMB2,製作了僅在單側腎臟誘導足細胞損傷(腎小球病,glomerulopathy)的2K1N小鼠模型。
當使用該模型僅對單側腎臟誘導損傷時,觀察到受損腎臟的腎血流呈時間依賴性下降,源自受損腎臟的蛋白尿消失。與此相對,另一側健康腎臟代償性地承擔起維持功能,全身的水鹽平衡和血壓得以維持並趨於穩定。
另一方面,在雙側腎臟受到同等損傷的2K2N模型中,未觀察到血流不均衡,全身性水腫和蛋白尿情況惡化。
研究團隊進一步使用2K1N模型,詳細調查腎臟內的基因表現及分子水平變化後發現,在受損腎臟和健康腎臟之間,與升高血壓的激素AngII生成相關的蛋白質水解酶「腎素Ren1」的表達存在明顯的左右差異,腎臟AngII的表達量也在受損腎臟一側顯著增加。
針對這一情況,施用血管收縮素轉換酶抑制劑後,血流下降和腎小球病灶得到改善,表明這種局部的AngII不均衡引起了受損腎臟的血流下降和腎小球衰竭。
研究發現,在左右腎臟試圖相互保持平衡的過程中,該平衡的崩潰與腎臟疾病的進展密切相關。
川西教授表示:「該研究始於筑波大學長田道夫教授(當時)的提議。當時還是研究生的坂本和雄成功完成了使用轉基因小鼠進行的高難度病灶腎臟移植,並由此發現了在正常腎臟和受損腎臟之間發生了資訊交流(串擾)。特別是,腎臟為保護身體而激活腎素-血管收縮素系統(RAS)的新功能得以闡明,目前研究已拓展到移植腎臟的領域。」
原文:《科學新聞》
翻譯:JST客觀日本編輯部
【論文資訊】
期刊:Proceedings of the Japan Academy, Series B
論文:A Novel Glomerulopathy Model Demonstrates Renal Counterbalance via Local Angiotensin II Regulation
DOI:10.2183/pjab.101.025